
HAlign
Download HAlign
latest release of HAlign 2: HAlign2.1.jar source code
latest release of HAlign 3: HAlign2.1.jar source code
We are now actively developing a new version. We strongly recommend you to try this version if you want to align nucleic acid sequences.
A series of detailed usage commands are showed in document part. Besides, a user-friendly server are ready now.
Development environments:- JDK 1.8
- Hadoop 2.7.2
- Spark 2.0.2
- Intellij IDEA (Maven)
Dataset
mitochondrial genomes- Ref: Tanaka M., et al. (2004) Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res,14(10a), 1832-1850.
- Download (zipped file): 1x (219KB) 20x (4.27MB) 50x (10.666MB) 100x (21.325MB)
- Ref: DeSantis, T. Z., et al.(2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res, 34, W394-399.
- Download (zipped file): small (21.864MB) big (197.224MB)
- Ref: Thompson, J.D., et al., BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins: Structure, Function, and Bioinformatics, 2005. 61(1): p. 127-136.
- Download (zipped file): 1x (4.16MB)
- Ref: Edgar, Robert C. Bench. 2009.
- Link: BENCH (20.4MB)
Related Tools
SP is a jar file, which can be used to compute the average value of sum-of-pair score for the alignment result.- jar: download
- source code
- Usage: java -jar sp.jar fasta_file [parameter.txt] [-s] where fasta_file is the alignment result, and parameter.txt stores the parameters(it can be omitted), and -s is the resample result(If there are too many sequences, -s can save the running time).
Document
Usages
1. Stand-alone mode
# java -jar HAlign2.1.jar <mode> <input-file> <output-file> <algorithm>
- mode:
-localMSA,-localTree. - input-file: local fasta format file, required.
- output-file: local fasta format file, just a file name, required.
-
algorithm: sequence alignment algorithms, required for
-localMSAmode, but none for-localTreemode.0represents the suffix tree algorithm, the fastest, but only for DNA/RNA;1represents the KBand algorithm based BLOSUM62 scoring matrix, only for Protein;2represents the KBand algorithm based on affine gap penalty, only for DNA/RNA;3represents the trie tree alignment algorithm, but slower and only for DNA/RNA;4represents the basic algorithm based the similarity matrix, the slowest and only for DNA/RNA. But it is the most accurate in the case of lower sequences similarity.
2. Hadoop cluster mode
# hadoop jar HAlign2.1.jar <mode> <input-file> <output-file> <algorithm>
- mode:
-hadoopMSA. - input_file/output_file/algorithm type: same as stand-alone mode.
3. Spark cluster mode
# spark-submit --class main HAlign2.1.jar <mode> <input-file> <output-file> <algorithm>
- mode:
-sparkMSA,-sparkTree. - input-file: local fasta format file, required.
- output-file: local fasta format file, just a file name, required.
- algorithm: sequence alignment algorithms, required for
-sparkMSAmode, but none for-sparkTreemode.0represents the suffix tree algorithm, the fastest, but only for DNA/RNA;1represents the KBand algorithm based BLOSUM62 scoring matrix, only for Protein.
Workflow of Trie Tree
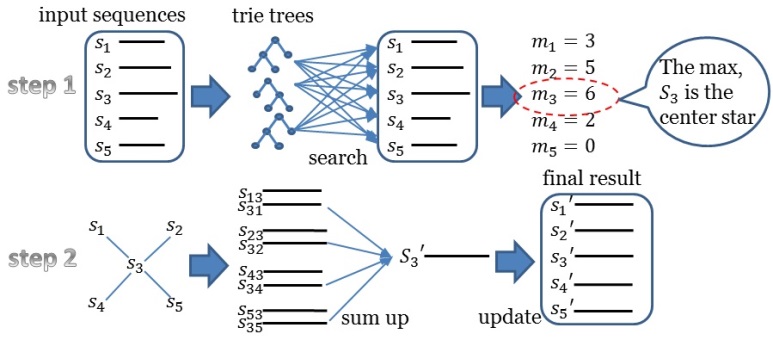
Fig. 1. Workflow of Trie Tree
As shown in the above figure, trie tree approach need to find the center star sequence first, then sum up the results for the final results. The Trie Tree Version runs quickly than the K-Band Version. However, the K-Band Version considers the Affine Gap Penalty while the Trie Tree Version didn't. K-Band approach can be applied for DNA, RNA and Protein multiple sequences alignment, which has a key role in multiple sequences alignment. More detailed information is shown in citation paper.Workflow of Hadoop Map Reduce
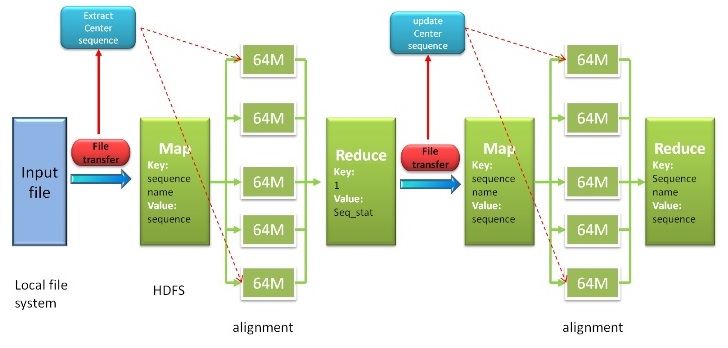
Fig. 2. Workflow of Hadoop Map Reduce
Based on map-reduce technique of hadoop, our program can work as hadoop mode for minimizing time cost. Our program reads input file from local file system, then be transformed into format key-value pair lines file and saved in hadoop distributed file system (HDFS). Next, pair sequences alignment procedure will be handed out, which is distributed parallel computing. Our program will search the unique center star sequence after this step, then next step will begin. After summing up procedure, reduce method will generate the final result. More detailed information is shown in citation paper.Server
Web server on the clusters in Tianjin University (recommended)
- Spark & Hadoop cluster
The cluster contains 12 nodes with high bandwidth. Each node has 94GB memory size and 2 CPUs (Intel(R) Xeon(R) CPU E5-2620 v3 @ 2.40GHz). - NVIDIA K80 GPU cluster new
The cluster contains 3 nodes with high bandwidth, including 2 K80 nodes and 1 MIC node. Each node has 504GB memory size and 2 CPUs (Intel(R) Xeon(R) CPU E5-2680 v4 @ 2.40GHz).
Web server on QingCloud platform
This website is deployed on the cluster in QingCloud. Before the experiments, you must startup hadoop cluster if they are dead, and time cost is about one minute. Besides, you can not use spark mode there.Citation
2. Shixiang Wan, Quan Zou. HAlign-II: efficient ultra-large multiple sequence alignment and phylogenetic tree reconstruction with distributed and parallel computing. Algorithms for Molecular Biology. 2017,12: 25.
3. Quan Zou, Shixiang Wan, Xiangxiang Zeng, Zhanshan Sam Ma. Reconstructing evolutionary trees in parallel for massive sequences. BMC System Biology. 2017, 11(Suppl 6): 100.
4. Wenhe Su, Xiangke Liao, Yutong Lu, Quan Zou, Shaoliang Peng. Multiple Sequence Alignment Based on a Suffix Tree and Center-Star Strategy: A Linear Method for Multiple Nucleotide Sequence Alignment on Spark Parallel Framework. Journal of Computational Biology. 2017, 24(12): 1230-1242.
5. Xi Chen, Chen Wang, Shanjiang Tang, Ce Yu, Quan Zou. CMSA: A heterogeneous CPU/GPU computing system for multiple similar RNA/DNA sequence alignment. BMC Bioinformatics. 2017, 18: 315.
Contact us
We thank to Mr. Yaozong Mao, who helped to develop the Hadoop version of our software tool. We thank to Mr. Shixiang Wan, who helped to developed the web server in Qingyun Hadoop System. The work was supported by the Natural Science Foundation of China (No.61370010).
Related Work
- MAFFT
- KAlign
- PASTA
- HAlign (Chinese version)
- HPTree
Copyright@2016 by Bioinformatics Laboratory, Tianjin University.