mAML: an automated machine learning pipeline with a microbiome repository for human disease classification
Note:
[numbers]
in square brackets are corresponding with the numbers marked on the figure in the right column
Set up parameters
The user can upload [2] BIOM file or Table file to start the pipeline. Press button [3] in the right column to preview the data uploaded. Transpose the feature matrix if the features are not in columns. As shown in the demo dataset[1], the microbiome data inputs should include the feature count/abundance/presence information for the samples, the metadata for the samples, and/or the metadata for the features. The features can represent OTUs as in 16S rRNA gene sequencing, genes as in metagenomics and transcriptomics, metabolites as in metabolomics, etc. The sample metadata should at least contain the labeling information of distinct groups, also called classes or phenotypes. Selection of different types of phenotype and the collapse of features into higher levels are supported.
The input features will be filtered at the default threshhold [4] (features lower than 20% within all classes will be disregarded).
Then, four feature subset selection (FSS) methods (the distal DBA method, HFE, Univariate feature selection and mRMR) were adopted to handle high-dimensional and sparse feature spaces as in microbiome datasets. Select None if you don't need to downsize the number of features.
The unbalanced datasets will be rebalanced [6] using SMOTE (Synthetic Minority Over-sampling Technique), ADASYN (Adaptive Synthetic Sampling Approach for Imbalanced Learning) or RandomOverSampler. Select None if the dataset is balanced.
Finally, the parameters or hyperparameters of the preprocessors [7] and classifiers [8, 10] could be edited and the adding and pruning of any preprocessor or classifier is supported. The grid search settings for the hyperparameters can be turned in the dict [9] function of each classifier.
By default, the pipeline will tune hyperparameters while searching [11], namely, select the best performing combination of preprocessors and non-tree based classifiers and the optimized hyperparameters for all classifiers at a time.
Before submission, the parameters of nested cross validation, the metric for model evaluation and the number of parallel process could be altered [12].
Fill in the email address and make sure all the above settings are checked. Then click "Confirmed parameter settings" [14] and press the power button below and the pipeline will run in the background. Check the latest information of the process in the right column [15].

Preview the result files and download
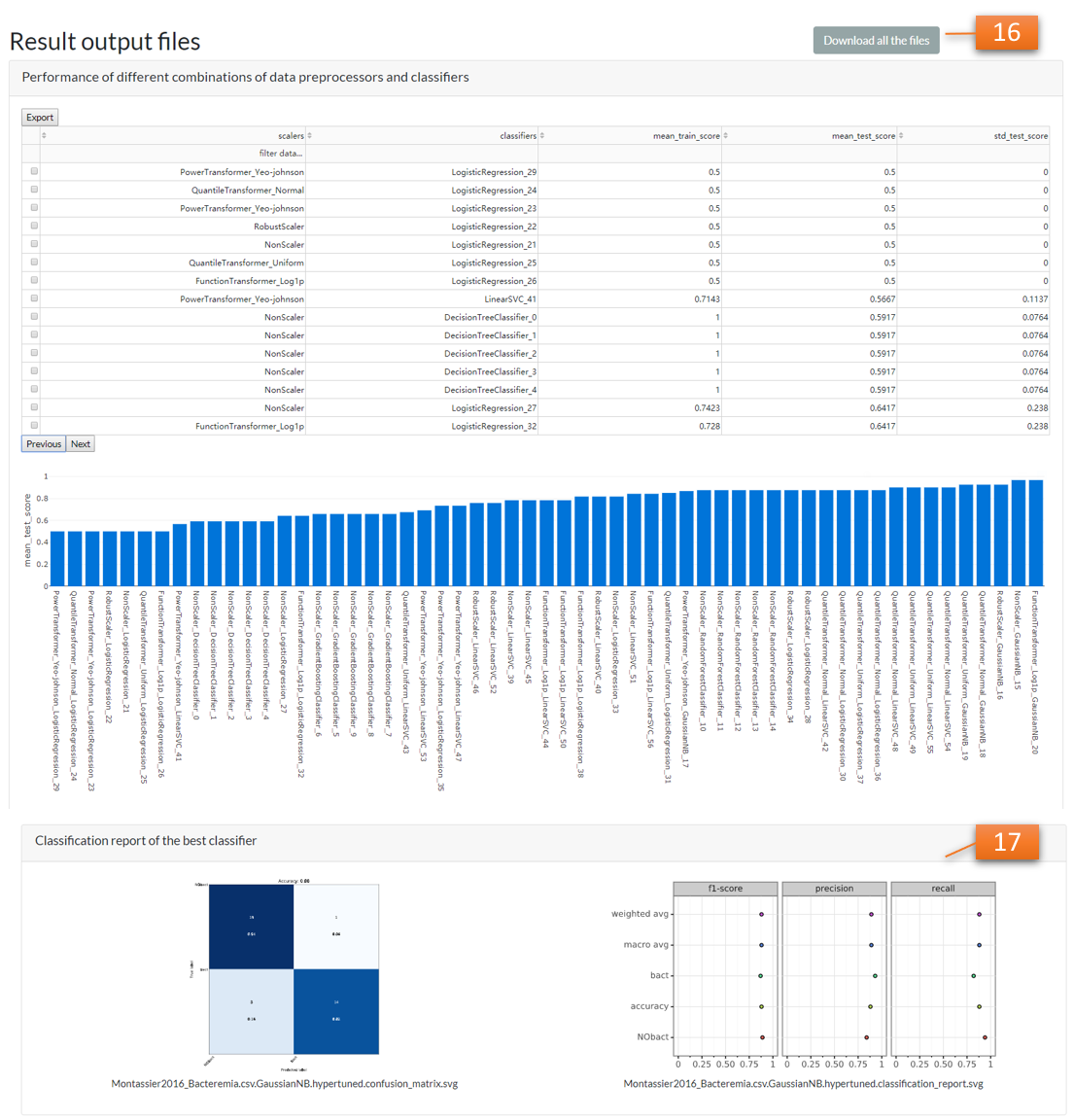
Once the run complete, all the result files will be compressed (downloadable [16]) and sent to the e-mail address [13] automatically. Preview the results and classification report [17] of the best model selected by the pipeline below.
Make predictions
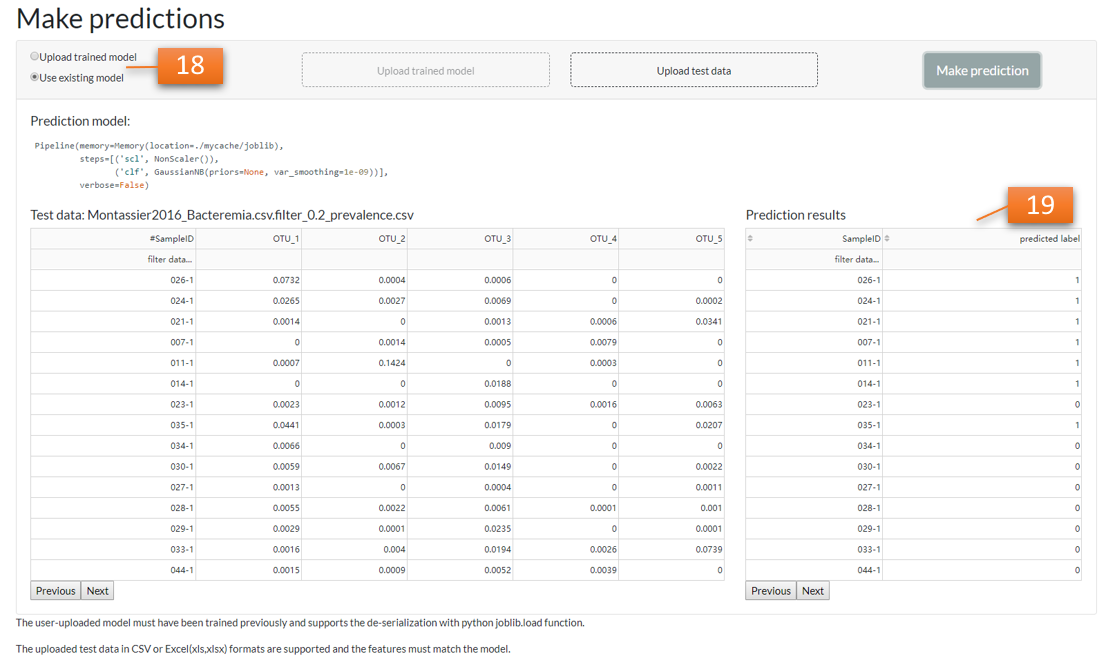
The user can upload new data to use the existing model, or upload previously trained model [18] to make new predictions [19].