Integral process
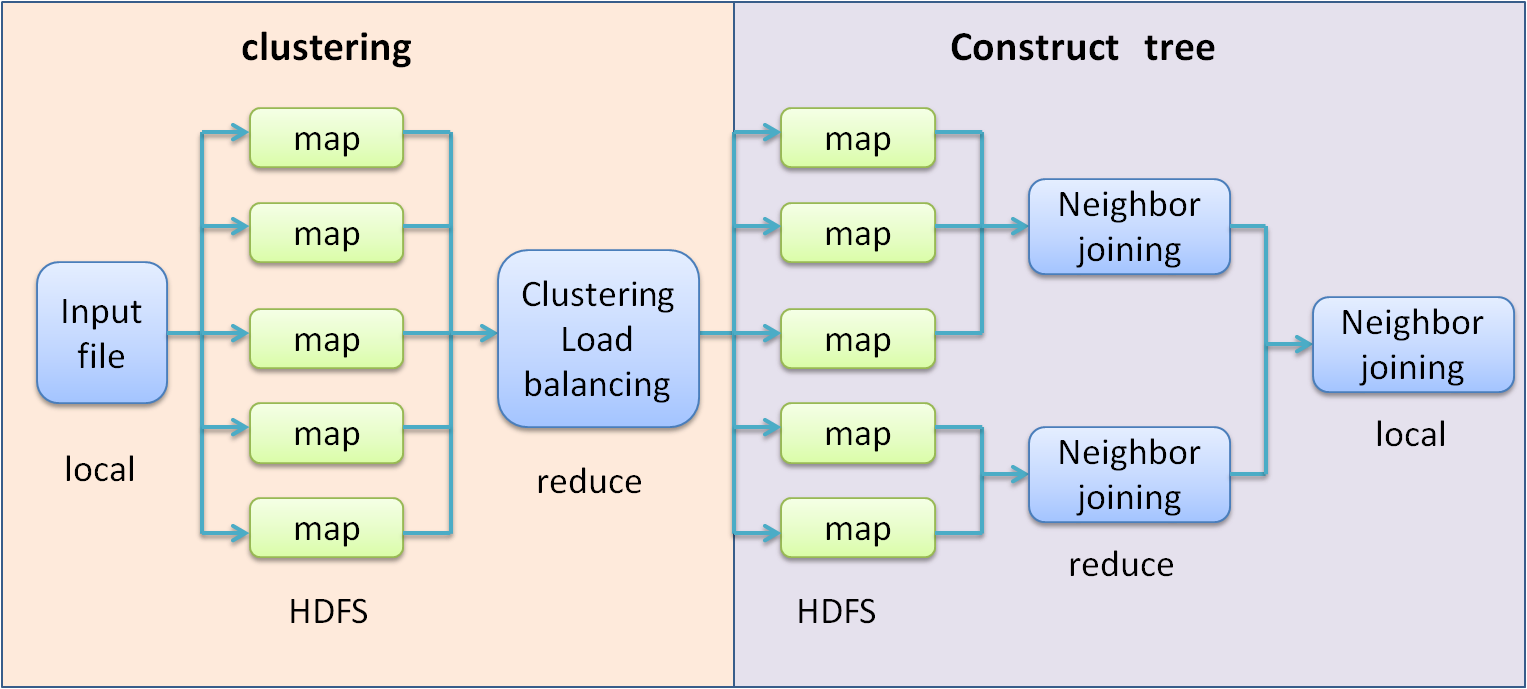
As the construct the phylogenetic tree can be regarded as a clustering process. We first clustering the data to some rough sets.
The clusters needn't too much precious .
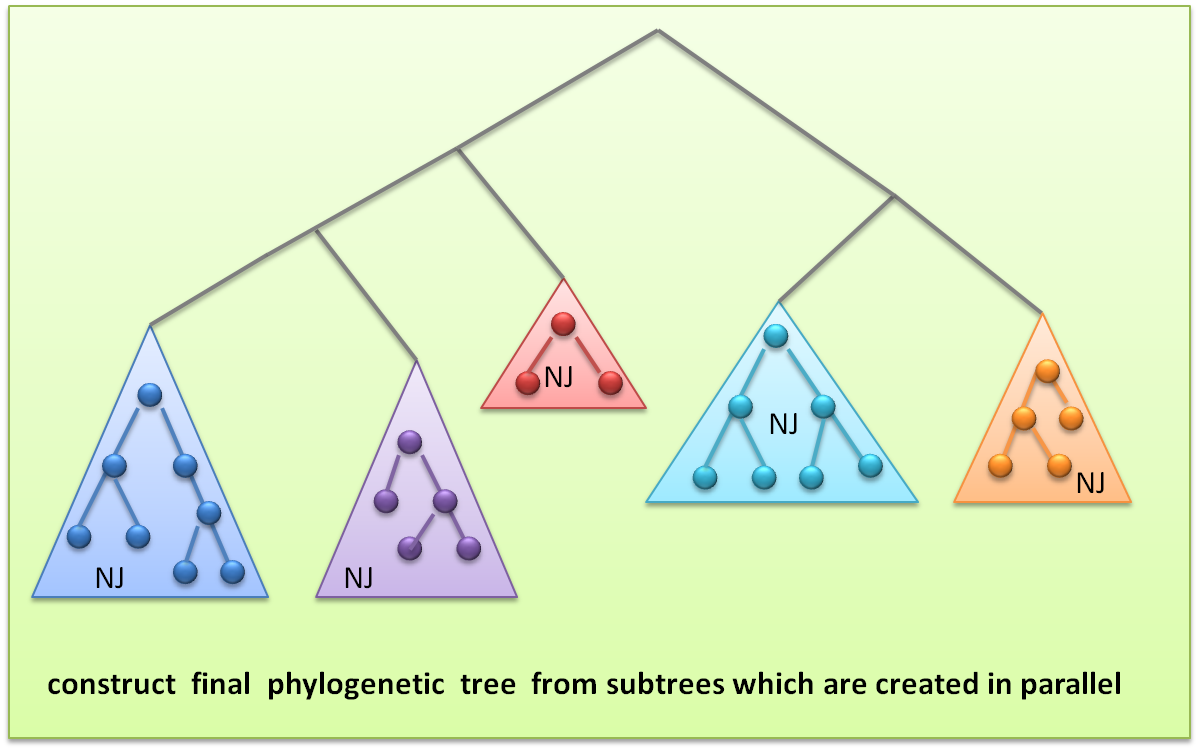
After the clustering , the Neighbor joining algorithm will execute in parallel on each sets.
After every sets construct the subtree , we select the root node as the representative of the subtree to construct the final phylogenetic tree.
Construct process
Download
Version 1.0. This version can accomplish the multiple sequence alignment process and construct phylogenetic tree process simultaneously.
Version 1.1. This version deals with the output of the MSA directly.
Manual
It runs in Linux OS with Hadoop platform.To execute the procedure we must complete the parameters file:
- local_path= the path that stores the input files
- dfs_path= the path in Hadoop file system
- filename= the input file name
- reduce_number= the max clusters number and it's also the maximum of reduce number
The parameters must be saved in the properties file , make sure the properties file and the jar file in the same directory . And then complete the follow steps:
| 1.start hadoop | # hadoop start-all.sh |
| 2.leave the safe mode | # hadoop dfsamdin -safemode leave |
| 3. start the program | # hadoop jar HPTree1.X.jar MSA_console properties_file |
The output files include
- dfs_path/MSAinput/inputKV The file stores the Key Value pairs.
- dfs_path/out/part-r-00000 The file stores sequence names, updated sequences, and the updated center star sequence.
- dfs_path/seqi/seqi The input file for the second Map function.
- dfs_path/theFinaloutput The final aligned sequence file
- local_path/MSAOutput The final aligned sequence file in local file system
- local_path/Phylogenetic_Output The final phylogenetic tree file in local file system
Notice
Please confirm that the input file is a DNA or RNA fasta file. Make sure Hadoop can work in your cluster.
All Rights Reserved Copyright @ 2015|Dr. Quan Zou
Last Modified in 2016/7/26